
Cosa è una interstiziopatia polmonare
L’interstiziopatia polmonare, chiamata anche fibrosi polmonare, è una alterazione del tessuto polmonare. Il termine è generico e può comprendere oltre 200 diverse malattie, che mostrano notevoli variazioni in termini di decorso clinico, trattamento e prognosi.
In generale, può dipendere da molte cause: alcune note (secondaria), altre invece sconosciute. In quest’ultimo caso si ha la cosiddetta fibrosi polmonare idiopatica (1).
L’interstiziopatia polmonare è caratterizzata da un’alterazione che riguarda gli “spazi” (interstizi) tra gli alveoli polmonari, che si ispessiscono e perdono elasticità. Questa condizione riduce la capacità degli alveoli di dilatarsi durante l’inspirazione, limitando gli scambi gassosi e l’ossigenazione del sangue provocando, di conseguenza, l’insufficienza respiratoria.
Le cause di interstiziopatia polmonare
Tra le molte cause di interstiziopatia polmonare troviamo:
- Malattie autoimmuni come l’artrite reumatoide o altre malattie autoimmuni del tessuto connettivo, che possono tradursi in fibrosi polmonari
- Alcuni farmaci, come l’amiodarone
- Alcuni chemioterapici, che possono determinare fibrosi polmonari
- Trattamenti radioterapici sul torace o sul mediastino, per esempio nel trattamento dei linfomi o delle neoplasie mammarie.
Più raramente, l’interstiziopatia può essere secondaria a infezioni polmonari tanto gravi da portare ad alterazione più o meno permanente del parenchima polmonare; l’infezione da coronavirus Covid-19, ad esempio, nei casi più gravi può alterare in modo permanente la struttura polmonare.
La forma più grave di interstiziopatia polmonare è la fibrosi polmonare idiopatica (2), una malattia di cui non si conoscono le cause, che porta a progressiva alterazione del parenchima polmonare, alla perdita della funzionalità del polmone, alla insufficienza respiratoria, fino alla morte. Può essere caratterizzata da lunghi periodi di stabilità e da improvvise accelerazioni.

Interstiziopatia polmonare – Diagnosi
Anamnesi nell’interstiziopatia polmonare
L’interstiziopatia non è generalmente una malattia acuta, per cui raramente si manifesta con sintomi iniziali eclatanti. Si può avere tosse, o mancanza di respiro progressiva (dispnea), fino all’insufficienza respiratoria cronica.
Esame obiettivo nell’interstiziopatia polmonare
A livello toracico l’obiettività polmonare evidenzia spesso rantoli crepitanti alle basi.
Esami strumentali nella diagnosi di interstiziopatia polmonare
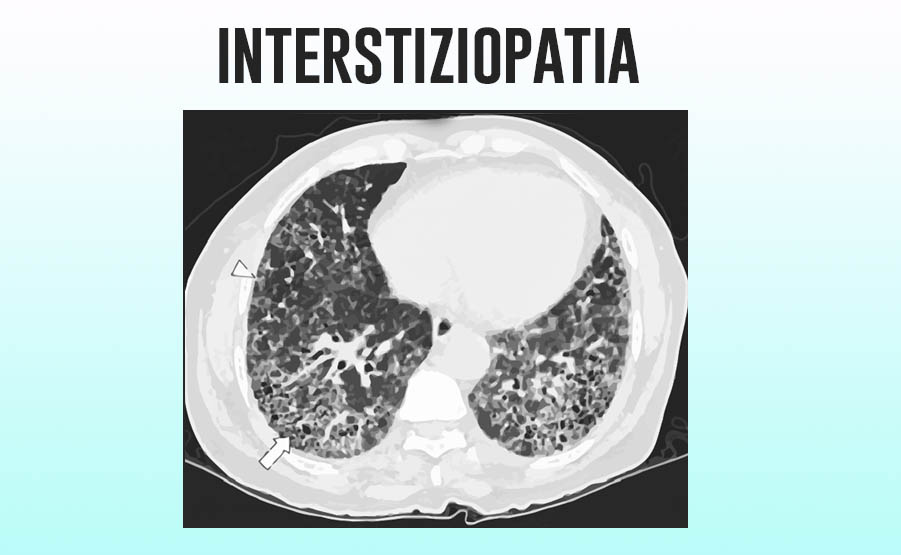
Il primo passo per la diagnosi di interstiziopatia polmonare è effettuare una TAC del torace ad alta risoluzione (HRCT).
La TC del torace infatti permette, in molti casi, di differenziare tra un tipo di interstiziopatia, denominata Fibrosi polmonare idiopatica (UIP – Usual Interstizial Pneumonia) e le altre forme di interstiziopatia polmonare.
In base alle caratteristiche radiologiche quindi, ma anche alla storia clinica del paziente (anamnesi), potremmo avere 4 tipi di “Pattern”:
- UIP
- Possible UIP
- Indeterminate UIP
- Interstiziopatia di altro tipo
Altre indagini utili sono:
- Esecuzione di una broncoscopia con lavaggio broncolaveolare (BAL): si tratta di un lavaggio profondo del polmone che permette di effettuare il conteggio delle sottopopolazioni cellulari presenti nel liquido di lavaggio e le confronta con le sottopopolazioni cellulari del sangue.
- Esecuzione dei markers di autoimmunità, indagini che insieme al quadro radiologico sono utili per escludere la presenza di altre cause di interstiziopatia.
La biopsia polmonare invece non è più una indagine di routine, viene effettuata solo nel caso sia necessario confermare la diagnosi di UIP in presenza di un quadro radiologico dubbio (indeterminate UIP), e solo se il paziente risulta candidato a terapia specifica.
La biopsia polmonare infatti non è quasi mai dirimente se effettuata tramite classica broncoscopia, ma è necessario effettuarla tramite criobiopsia, procedura che si fa in anestesia generale con un broncoscopio rigido, o tramite vero e proprio intervento chirurgico in videotoracoscopia.
La spirometria nella valutazione funzionale dell’interstiziopatia
Oltre alla diagnosi della tipologia di interstiziopatia, che si giova dell’immagine TC, è importante valutare qual è il grado di compromissione funzionale. Per ottenere questa informazione si utilizza la spirometria, che è in grado di misurare i volumi polmonari e quindi stimare il grado di danno funzionale in corso.
Quali sono le caratteristiche radiologiche della fibrosi polmonare idiopatica ( UIP)?
Il pattern della fibrosi polmonare idiopatica è caratterizzato dalla presenza di fibrosi nella zona subito al di sotto della pleura (subpleurica), con una distribuzione non omogenea nei due polmoni e che va aumentando dall’apice del polmone verso le basi. Negli stadi più avanzati sono presenti alterazioni importanti del parenchima polmonare come con le bronchiectasie, e un particolare aspetto radiologico chiamato “honey combing” (aspetto simile alle celle di un alveare).
Queste caratteristiche distinguono la fibrosi polmonare idiopatica (UIP) dalle altre forme di fibrosi, che mostrano invece altri aspetti radiologici; talvolta, tuttavia, anche in altre forme di fibrosi lo stravolgimento del parenchima polmonare può essere tale da rendere difficile distinguere una forma dall’altra.
Perché è importante distinguere le UIP dalle altre forme di fibrosi polmonare?
La UIP è una malattia grave e potenzialmente mortale, nelle sue fasi finali può portare a insufficienza respiratoria; può subire delle accelerazioni, dovute ad un infezione bronchiale o causate da una indagine diagnostica invasiva, ad esempio la biopsia. La UIP tuttavia, ad oggi ha anche la possibilità di avere un trattamento.
Esistono infatti due farmaci antifibrotici: il pirfenidone e il nintedanib, che hanno dimostrato essere utili nel rallentare il declino della funzionalità polmonare, che viene valutata tramite la spirometria e la misura della diffusione alveolo capillare del CO.
Quest’ultima in particolare ci dà la misura di quanto sia alterato/stravolto il parenchima polmonare: più è ridotta più il parenchima polmonare è alterato.
I farmaci antifibrotici a nostra disposizione devono essere somministrati, però, solo nel caso ci sia una elevata probabilità che il quadro radiologico/anamnestico sia compatibile con IP, in quanto devono essere assunti per lungo periodo e possono avere importanti effetti collaterali.
Per questo motivo l’inizio della terapia con tali farmaci viene sancito dopo una riunione multidisciplinare, a cui partecipano diversi specialisti: lo pneumologo, il radiologo, il reumatologo. La loro prescrizione è soggetta a piano terapeutico e devono essere effettuati stretti controlli degli esami ematochimici e clinici per il rinnovo del piano stesso.

Se ti è piaciuto questo articolo e vuoi rimanere aggiornato sulle nostre prossime pubblicazioni clicca qui per iscriverti alla nostra newsletter.
Fonti e note:
- Mikolasch TA, Garthwaite HS, Porter JC. Update in diagnosis and management of interstitial lung disease.
- Ganesh Raghu, martine Remy-Jardin, Jeffrey L. Diagnosis of Idiopathic Pulmonary Fibrosis. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline.